
Regulatory bodies in healthcare: an introduction
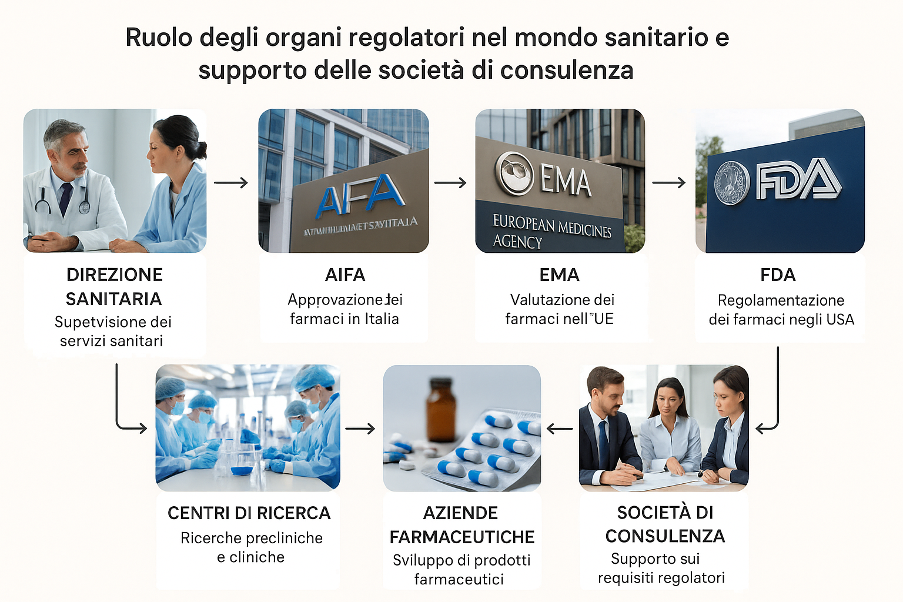
Regulatory bodies regulatory bodies in the healthcare sector play a key role in ensuring the safety, efficacy and quality of healthcare products and services. Government bodies, the Ministry of Health and other related Ministries, the Istituto Superiore di Sanità, and related Agencies, such as the Agenzia Italiana del Farmaco (AIFA) in Italy, the EMA for the whole EU, are in charge of overseeing the entire lifecycle of medicines and medical devices from design to market, as well as the underlying clinical research processes.
Their main mission è protecting public health. With the various European Directives issued over the past two decades, a number of areas have been innovated that regulate clinical research activities and the way medicines and medical devices are developed and marketed, aiming more and more at enhancing patient safety and improving the underlying operational processes.
Their main mission is to protect public health.
The challenges and complexity of regulating a research centre
Health regulation is a complex, dynamic and constantly evolving process. On the one hand, Regulatory Agencies must navigate an environment characterised by rapid technological progress, rising patient expectations and increasing globalisation. On the other hand, they have to balance the need to promote innovation with the imperative to guarantee safety and control health system costs. Their ability to keep up with new scientific breakthroughs, such as gene therapies or personalised medicine, is crucial for the timely availability of innovative treatments on the market. On the other hand, manufacturing companies, especially large ones, need to be able to enter markets quickly after years of research for a return on their investments and also have to deal with a market that is subject to price rules negotiated with the pharmaceutical companies themselves.
The stringent regulations on the market for pharmaceuticals, such as gene therapies or personalised medicine, are crucial for the timely availability of innovative treatments on the market.
Strict regulations governing the research phase, the bureaucratic procedures for access to the production of dossiers prior to market authorisation, price negotiation, market access policies, as well as post-sale risk monitoring systems require increasingly structured organisations; structured organisations, with very high and technically qualified professional skill profiles, as well as organisational models to manage the individual stages from R&S to market share conquest.
The Research Centres authorised to process “mogm” and the authorisation and management implications.
The evolution of biotechnology has brought to the attention of health and scientific institutions the need to define clear rules regarding the genetic manipulation and use of genetically modified microorganisms (GMM). These organisms, derived from genetic engineering processes, represent essential tools for clinical, pre-clinical and applied research, but at the same time have major implications in terms of biosafety, traceability and regulatory management.
Italy, in line with European Union directives, has developed a complex regulatory framework governing the authorisation of research centres for the treatment of GMOs, imposing stringent risk assessment criteria, infrastructure requirements and management responsibilities. This article intends to analyse the role of authorised centres, the main procedural steps for obtaining the ministerial green light, and the managerial and operational repercussions of their activities.
The regulation of OMCs in the field of research and development
The regulation of OMCMs lies at the crossroads between European regulations and national legislation. The main regulatory references are:
- Directive 2009/41/EC on the contained use of genetically modified micro-organisms, which establishes common principles of risk assessment and classification of activities.
- Legislative Decree 206/2001, which transposes EU legislation and regulates the conditions of authorisation and supervision of centres wishing to operate with GMOs.
- Ministerial Guidelines and Measures of the Istituto Superiore di Sanità (ISS), which provide technical–operational criteria and health surveillance modalities for the operators involved.
In this framework, research centres can operate only after obtaining a formal authorisation from the Ministry of Health, following the opinion of the Interministerial Commission for Biosafety and Biotechnology and the technical–scientific evaluation of the ISS.
To be authorised, a research centre must demonstrate that it meets a number of structural, organisational and scientific requirements. Among the main ones:
1. Suitable infrastructures:
- Rooms and laboratories adequately designed to confine biohazard.
- Ventilation and air filtration systems, controlled access, differentiated routes for personnel and materials.
- Presence of bio-waste decontamination and disposal areas.
2. Internal organisation:
- Appointment of a Biosafety Manager, with certified competence.
- Adoption of risk management manuals and standard operating protocols.
- Continuous training of technical and scientific staff.
3. Approval procedures:
- Pre-assessment of the risk (Risk Assessment) of each research project.
- Communication to the Ministry of Health by means of the so-called “notification of use”.
- Approval conditional on the’adoption of containment measures appropriate to the identified class of risk.
Management implications
Once authorised, research centres are faced with a series of management responsibilities that go beyond mere formal compliance with regulations.
1. Biosafety and health surveillance.
The protection of personnel and the environment is a priority. Operators are subjected to:
- periodic health surveillance programmes, in cooperation with the competent doctors;
- environmental monitoring and microbiological controls;
- periodic simulations of the management of accidents or accidental contamination.
2. Traceability and documentation
Every handling of MOGM must be recorded and stored in dedicated archives to ensure traceability of samples, procedures and any controlled releases. Documentation is subject to ministerial audits and surprise inspections.
3. Legal and ethical responsibility
The management of the centre and the scientific managers are personally liable for the activities carried out. Violations of the regulations may lead to suspension or revocation of the authorisation, as well as criminal consequences in the event of damage to persons or the environment.
4. Scientific and regulatory update
The evolution of biotechnology requires constant updating in both scientific and legislative fields. Centres must periodically adapt their procedures and promptly notify the Ministry of any infrastructural or design changes.
The strategic role of authorised centres
Criticism and future prospects
In spite of the solid regulatory framework, some criticalities emerge:
- Long authorisation times, which may slow down the start of clinical trials.
- Heterogeneity of interpretation between different regions or institutions.
- High management costs, related to the maintenance of biosafety standards.
Looking ahead, it appears necessary to simplify certain procedures, to favour platforms for sharing between centres through a hub & spoke network model, in order to optimise management costs and at the same time develop an increase in research and development skills, and also to invest in specialised training. The aim is to ensure both security and competitiveness, allowing Italy to position itself as an attractive location for advanced biotechnological research.
In summary, research centres authorised to process GMOs represent crucial infrastructures for scientific and therapeutic progress, and a powerful development asset for Italy, in a highly competitive market of high scientific and technological knowledge. Their activity, strictly regulated and monitored, entails a complex system of authorisation and management responsibilities. On the one hand, this ensures the protection of public health and the environment; on the other, it imposes on the research organisations a continuous commitment in terms of compliance, innovation and updating.
In short, ministerial authorisation is not only a constraint, but also a certification of excellence, enabling the centres to play a key role in the development of new frontiers in medicine and biotechnology.
They offer indispensable support in dealing with regulatory complexities and navigating through bureaucratic procedures, and support research organisations and manufacturers in defining successful strategies for bringing new discoveries to market.
Their areas of expertise include:
- Regulatory consulting: they help companies understand the requirements for approval of drugs and medical devices, supporting them in the preparation and submission of registration dossiers. They therefore work in understanding the most effective ways of submitting innovative products, also assessing the characteristics of competing products, and helping companies to identify the best price and cost-effectiveness strategy for submitting dossiers.
- They also help companies to identify the most effective way of submitting dossiers.
- Strategy and market access: they provide advice on how to position a product in the market, considering pricing dynamics, reimbursement and health authority decisions. They also support the introduction of innovative strategies to find the right balance between sales and purchasing strategies by healthcare companies.
- Compliance and quality: they assist companies in setting and maintaining quality standards in terms of layouts, technologies, safety systems and the organisational and management model underlying the R&S processes, and in supporting entities in preparing for inspections by regulatory bodies.
- Project Management and Clinical Trials: they support the planning and execution of clinical trials in compliance with current regulations, operating as staff members of the entities or companies in individual clinical research projects, supporting the appropriate organisation of the various teams of researchers, acting as a liaison between scientific project management and Company or Entity management, offering project management methodologies, to ensure compliance with objectives, consistently with the financial resources and timeframes set.
Consulting companies, with their in-depth knowledge of regulations and procedures, act as a bridge between companies and regulatory bodies. They help to smoothen internal processes when the management of the regulatory process for example is not ècore, to reduce approval times and to minimise the risks of non-compliance; and also to assess the new impacts that new technologies may have on the patient experience, or client experience.
A final example on which the consultancy companies are proceeding is that of supporting Research Organisations and manufacturers of medical devices for remote monitoring and public and private healthcare institutions in the introduction of technologies that impact on patients' everyday life, highlighting how, for example, a simple telemonitoring of certain parameters requires an organisational set-up of both healthcare profiles and customer experience technicians and services to be developed and strengthened.
The consultancy companies are also working with the medical device manufacturers to develop and implement new technologies that impact on patients' daily lives.
The opportunity for consultancy companies to be able to act in parallel on different industrial situations also allows the exchange of best practices, enabling research organisations and manufacturers to act more quickly and reducing possible risks of delays in market entry.
This way, support for the development of new products and services is also an important part of the project.
In this way, they support industry in bringing new drugs and innovative technologies to market more efficiently, indirectly contributing to patient welfare.
The Principal Investigator's Perspective
From the perspective of a Principal Investigator involved in the conduct of clinical trials, the aspects offered by consultancy companies take on different importance depending on the phases and responsibilities of research. The project management phase of clinical trials is essential to ensure proper planning, team organisation and compliance with regulatory and contractual deadlines. The methodological governance provided is often decisive for the success of a trial. Regulatory consultancy is essential to accurately address the complexity of approval procedures and to reduce the risk of delays or non-compliance.
Accompanying these two phases is the Conformity and Quality Assessment, which is important to ensure that processes meet international standards, guaranteeing safety and credibility in the eyes of regulatory authorities and stakeholders.
Last but not least, the Market Access Strategy is indispensable to orientate the choices for possible future positioning of research results, with a direct impact on the possibilities of technology transfer and the attractiveness of studies for sponsors and institutions.
In summary, for a Principal Investigator, the ability of consultancy companies to support scientific activities with management, regulatory and strategic competences represents an essential added value, especially in a competitive and highly complex context such as that of modern clinical research.
Conclusions
In conclusion, regulatory agencies and consulting firms, while having distinct roles and responsibilities, work in an interconnected ecosystem. Regulatory agencies set the rules of the game, while consulting firms help companies to play it to the best of their ability. This collaboration, albeit indirect, is crucial for the efficiency and sustainability of the healthcare system, ensuring that patients have access to safe and effective therapies in the shortest possible time, and for the healthcare system to be able to optimise the processes and economies underlying the introduction of continuous technological innovations. The integration of knowledge and the possibility of a multidisciplinary scenario finds its place in the medical management of hospitals.
Regulatory and Reference Bibliography
- Directive 2009/41/EC of the European Parliament and of the Council of 6 May 2009 on the contained use of genetically modified micro-organisms
- .
- Legislative Decree No. 206 – 12 April 2001; Implementation of Directive 98/81/EC on the contained use of genetically modified micro-organisms.
- Guidelines of the Ministry of Health on the contained use of genetically modified micro-organisms and risk assessment criteria.
- Documents and circulars of the Istituto Superiore di Sanità (ISS) on biosafety and biotechnologies.
- National Committee for Biosafety, Biotechnology and Life Sciences (CNBBSV) – Reports and Recommendations.


