

Organi regolatori nel settore sanitario: un’introduzione
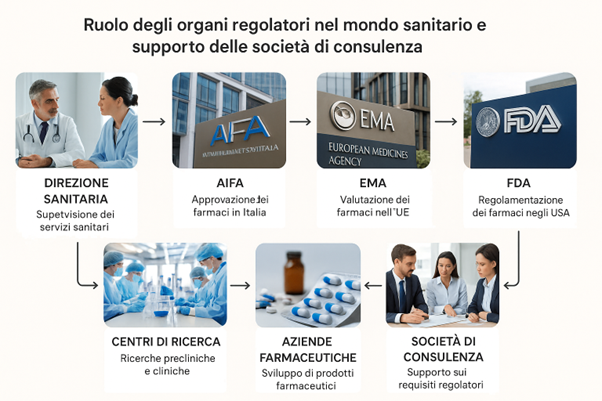
Gli organi regolatori nel settore sanitario svolgono un ruolo fondamentale nel garantire la sicurezza, l’efficacia e la qualità dei prodotti e dei servizi sanitari. Gli enti governativi, Ministero della Salute e altri Ministeri correlati, l’Istituto Superiore di Sanità, e le relative Agenzie, come l’Agenzia Italiana del Farmaco (AIFA) in Italia, l’EMA per tutta l’UE, hanno il compito di supervisionare l’intero ciclo di vita dei farmaci e dei dispositivi medici dalla progettazione alla immissione sul mercato, nonché i relativi processi di ricerca clinica sottostanti.
La loro missione principale è proteggere la salute pubblica. Con le varie Direttive Europee, emanate nel corso degli ultimi vent’ anni, sono stati innovati alcuni ambiti che regolamentano le attività di ricerca clinica, e le modalità di sviluppo e la messa in commercio dei farmaci e dispositivi medici, puntando sempre di più a potenziare la sicurezza del paziente, e a migliorare i processi operativi sottostanti.
Le sfide e la complessità della regolamentazione di un centro di ricerca
La regolamentazione in campo sanitario è un processo complesso, dinamico e in continua evoluzione. Da un lato, le Agenzie Regolatorie devono navigare in un ambiente caratterizzato da un rapido progresso tecnologico, un aumento delle aspettative dei pazienti e una crescente globalizzazione. Inoltre, devono bilanciare la necessità di promuovere l’innovazione con l’imperativo di garantire la sicurezza e il controllo dei costi di sistema sanitario. La loro capacità di rimanere al passo con le nuove scoperte scientifiche, come le terapie geniche o la medicina personalizzata, è cruciale per la tempestiva disponibilità di trattamenti innovativi sul mercato. Dall’altro le aziende produttrici, soprattutto quelle di grandi dimensioni necessitano dopo anni di ricerca, di poter entrare nei mercati rapidamente per il ritorno dagli investimenti e devono anche confrontarsi con un mercato che per quanto riguarda i farmaci sottostà a regole di prezzo negoziate con gli Enti stessi.
Le stringenti norme che regolano la fase di ricerca, gli iter burocratici per accedere alla produzione dei dossier preliminari alla autorizzazione al mercato, la negoziazione dei prezzi, le politiche di market access, nonché i sistemi di monitoraggio dei rischi post vendita richiedono organizzazioni sempre più strutturate, con profili di competenze professionali molto alte e tecnicamente qualificate, nonché modelli organizzativi deputati a gestire le singole fasi dalla R&S alla conquista delle quote di mercato.
I Centri di Ricerca autorizzati al trattamento dei “mogm” e le implicazioni autorizzative e gestionali.
L’evoluzione delle biotecnologie ha posto all’attenzione delle istituzioni sanitarie e scientifiche la necessità di definire regole chiare in materia di manipolazione genetica e utilizzo di microrganismi geneticamente modificati (MOGM). Tali organismi, derivati da processi di ingegneria genetica, rappresentano strumenti essenziali per la ricerca clinica, preclinica e applicata, ma comportano al contempo rilevanti implicazioni in termini di biosicurezza, tracciabilità e gestione regolatoria.
L’Italia, in linea con le direttive dell’Unione Europea, ha sviluppato un articolato quadro normativo che disciplina l’autorizzazione dei centri di ricerca destinati al trattamento dei MOGM, imponendo criteri stringenti di valutazione del rischio, requisiti infrastrutturali e responsabilità gestionali. Questo articolo intende analizzare il ruolo dei centri autorizzati, le principali fasi procedurali per l’ottenimento del via libera ministeriale e le ricadute gestionali e operative legate alla loro attività.
La disciplina dei MOGM si colloca all’incrocio tra normativa europea e legislazione nazionale. I principali riferimenti normativi sono:
– Direttiva 2009/41/CE sull’impiego confinato di microrganismi geneticamente modificati, che stabilisce principi comuni di valutazione del rischio e di classificazione delle attività.
– Decreto Legislativo 206/2001, che recepisce la normativa comunitaria e disciplina le condizioni di autorizzazione e vigilanza dei centri che intendono operare con MOGM.
– Linee guida ministeriali e provvedimenti dell’Istituto Superiore di Sanità (ISS), che forniscono criteri tecnico–operativi e modalità di sorveglianza sanitaria per gli operatori coinvolti.
In questo quadro, i centri di ricerca possono operare solo dopo aver ottenuto un’autorizzazione formale dal Ministero della Salute, previo parere della Commissione interministeriale per la biosicurezza e le biotecnologie e valutazione tecnico–scientifica dell’ISS.
Per essere autorizzato, un centro di ricerca deve dimostrare il possesso di una serie di requisiti strutturali, organizzativi e scientifici. Tra i principali:
1. Infrastrutture idonee:
– Locali e laboratori adeguatamente progettati per confinare il rischio biologico.
– Sistemi di ventilazione e filtrazione dell’aria, accessi controllati, percorsi differenziati per personale e materiali.
– Presenza di aree di decontaminazione e smaltimento rifiuti biologici.
2. Organizzazione interna:
– Nomina di un Responsabile della Biosicurezza, con competenze certificate.
– Adozione di manuali di gestione del rischio e protocolli operativi standard.
– Formazione continua del personale tecnico e scientifico.
3. Procedure autorizzative:
– Valutazione preventiva del rischio (Risk Assessment) di ciascun progetto di ricerca.
– Comunicazione al Ministero della Salute mediante la cosiddetta “notifica di impiego”.
– Approvazione condizionata all’adozione di misure di contenimento adeguate alla classe di rischio identificata.
Implicazioni gestionali
Una volta autorizzati, i centri di ricerca devono affrontare una serie di responsabilità gestionali che vanno oltre la semplice osservanza formale delle norme.
1. Biosicurezza e sorveglianza sanitaria.
La tutela del personale e dell’ambiente è prioritaria. Gli operatori sono sottoposti a:
– programmi di sorveglianza sanitaria periodica, in collaborazione con i medici competenti;
– monitoraggi ambientali e controlli microbiologici;
– simulazioni periodiche di gestione di incidenti o contaminazioni accidentali.
2. Tracciabilità e documentazione
Ogni manipolazione di MOGM deve essere registrata e conservata in archivi dedicati, per garantire la rintracciabilità dei campioni, delle procedure e delle eventuali emissioni controllate. La documentazione è soggetta a verifiche ministeriali e ispezioni a sorpresa.
3. Responsabilità legale ed etica
La direzione del centro e i responsabili scientifici rispondono personalmente delle attività svolte. Eventuali violazioni delle norme possono determinare la sospensione o revoca dell’autorizzazione, oltre a conseguenze penali in caso di danni a persone o ambiente.
4. Aggiornamento scientifico e normativo
L’evoluzione delle biotecnologie impone un costante aggiornamento sia in ambito scientifico sia legislativo. I centri devono adeguare periodicamente le proprie procedure e comunicare tempestivamente al Ministero eventuali modifiche infrastrutturali o progettuali.
Il ruolo strategico dei centri autorizzati
Ottenere l’autorizzazione al trattamento dei MOGM non è soltanto un adempimento formale, ma rappresenta un riconoscimento di competenza scientifica e affidabilità gestionale. I centri autorizzati assumono un ruolo strategico in diversi ambiti:
– Ricerca clinica di fase 1 e 2: sviluppo di nuove terapie avanzate, comprese le terapie geniche e cellulari.
– Ricerca traslazionale: collegamento tra risultati di laboratorio e applicazioni cliniche.
– Collaborazioni internazionali: partecipazione a network di ricerca europei e globali, con accesso a finanziamenti competitivi.
– Innovazione industriale: trasferimento tecnologico verso imprese biotech e farmaceutiche.
Criticità e prospettive future
Nonostante l’impianto normativo solido, emergono alcune criticità:
– Tempi autorizzativi lunghi, che possono rallentare l’avvio di studi clinici.
– Eterogeneità di interpretazione tra diverse regioni o istituzioni.
– Alti costi gestionali, legati al mantenimento degli standard di biosicurezza.
In prospettiva, appare necessario semplificare alcune procedure, favorire piattaforme di condivisione tra centri attraverso un modello di rete Hub & Spoke, al fine di ottimizzare i costi di gestione e contestualmente sviluppare una crescita delle competenze in ricerca e sviluppo, e inoltre, investire in formazione specialistica. L’obiettivo è garantire al contempo sicurezza e competitività, permettendo all’Italia di posizionarsi come polo attrattivo per la ricerca biotecnologica avanzata.
In sintesi, i centri di ricerca autorizzati al trattamento dei MOGM rappresentano infrastrutture cruciali per il progresso scientifico e terapeutico, ed un potente asset di sviluppo per l’Italia, in un mercato fortemente competitivo ad alta conoscenza scientifica e tecnologica. La loro attività, strettamente regolata e monitorata, comporta un complesso sistema di responsabilità autorizzative e gestionali. Da un lato, ciò assicura la protezione della salute pubblica e dell’ambiente; dall’altro, impone agli enti di ricerca un continuo impegno in termini di conformità, innovazione e aggiornamento.
In definitiva, l’autorizzazione ministeriale costituisce non solo un vincolo, ma anche una certificazione di eccellenza, che consente ai centri di svolgere un ruolo chiave nello sviluppo di nuove frontiere della medicina e della biotecnologia.
Il ruolo delle società di consulenza
Le società di consulenza si inseriscono in questo contesto come partner strategici per le aziende e gli enti pubblici e privati che operano nella ricerca e nello sviluppo innovativo del settore sanitario.
Offrono un supporto indispensabile per affrontare le complessità normative e per navigare tra le procedure burocratiche, e supportano gli enti di ricerca, le aziende produttrici, anche nella definizione delle strategie di successo per introdurre le nuove scoperte sul mercato.
Le loro aree di competenza includono:
- Consulenza regolatoria: aiutano le aziende a comprendere i requisiti necessari per l’approvazione di farmaci e dispositivi medici, supportandole nella preparazione e sottomissione dei dossier di registrazione. Operano pertanto nel comprendere le modalità più efficaci di presentazione de prodotto innovativo valutando anche le caratteristiche dei prodotti concorrenti, e aiutando le aziende a individuare la modalità di presentazione dei dossier per la migliore strategia di prezzo e di costo efficacia.
- Strategia e market access: forniscono consulenza su come posizionare un prodotto sul mercato, considerando le dinamiche dei prezzi, i rimborsi e le decisioni delle autorità sanitarie. Sono di supporto anche per introdurre strategie innovative che consentono di individuare il giusto equilibrio tra strategie di vendita e di acquisto da parte delle aziende sanitarie.
- Conformità e qualità: assistono le aziende nell’allestimento e nel mantenimento degli standard di qualità dal punto di vista dei layout, delle tecnologie, dei sistemi di sicurezza e del modello organizzativo e gestionale sottostante i processi di R&S, e nel supportare gli enti alla preparazione per ispezioni da parte degli organi regolatori.
- Project Management e Studi clinici: supportano la pianificazione e l’esecuzione di studi clinici conformi alle normative vigenti, operando come operatori in staff agli enti o aziende nei singoli progetti di ricerca clinica, supportando una adeguata organizzazione dei vari team di ricercatori, facendo da raccordo tra direzione scientifica di progetto e Direzione Aziendale o di Ente, offrendo metodologie di governo progettuale, per garantire il rispetto degli obiettivi, coerentemente con le risorse finanziarie e i tempi prefissati.
Le società di consulenza, con la loro profonda conoscenza delle normative e delle procedure, agiscono come un ponte tra le aziende e gli organi regolatori. Contribuiscono a snellire i processi interni quando la gestione del processo regolatorio ad esempio non è core, a ridurre i tempi di approvazione e a minimizzare i rischi di non conformità e a valutare anche i nuovi impatti che le nuove tecnologie potranno avere sulla patient experience, o client experience.
Un esempio ultimo su cui le Società di consulenza stanno procedendo è quello di affiancare gli Enti di Ricerca e i produttori di dispositivi medicali di monitoraggio da remoto e gli enti sanitari pubblici e privati, nella introduzione di tecnologie che impattano sulla quotidianità dei pazienti, evidenziando come ad esempio un semplice telemonitoraggio di alcuni parametri richieda una assetto organizzativo sia di profili sanitari, sia di tecnici e servizi di Customer experience da sviluppare e potenziare.
L’opportunità per le società di consulenza di poter agire parallelamente su differenti situazioni industriali consente anche lo scambio delle best practice consentendo agli enti di ricerca e ai produttori di agire più rapidamente e riducendo possibili rischi di ritardi nell’ingresso sul mercato.
In questo modo, supportano l’industria nel portare sul mercato in modo più efficiente i nuovi farmaci e le tecnologie innovative, contribuendo indirettamente al benessere dei pazienti.
La prospettiva del Principal Investigator
Dal punto di vista di un Principal Investigator coinvolto nella conduzione di trial clinici, gli aspetti offerti dalle società di consulenza assumono un’importanza diversa a seconda delle fasi e delle responsabilità di ricerca. La fase di Project Management degli Studi clinici è fondamentale per garantire la corretta pianificazione, l’organizzazione dei team e il rispetto delle tempistiche regolatorie e contrattuali. La governance metodologica fornita è spesso determinante per il successo di un trial. La Consulenza regolatoria è essenziale per affrontare in modo accurato la complessità delle procedure di approvazione e per ridurre i rischi di ritardi o di mancata conformità.
Ad accompagnare queste due fasi, c’è anche la valutazione della Conformità e Qualità, importante per assicurare che i processi rispettino standard internazionali, garantendo sicurezza e credibilità agli occhi delle autorità regolatorie e degli stakeholder coinvolti.
Ultima, ma non meno importante è la Strategia di Market Access che è indispensabile per orientare le scelte relative ad eventuali posizionamenti futuri dei risultati della ricerca, con impatto diretto sulle possibilità di trasferimento tecnologico e sull’attrattività degli studi verso sponsor e istituzioni.
In sintesi, per un Principal Investigator, la capacità delle società di consulenza di affiancare l’attività scientifica con competenze gestionali, regolatorie e strategiche rappresenta un valore aggiunto imprescindibile, soprattutto in un contesto competitivo e ad alta complessità come quello della ricerca clinica moderna.
Conclusioni
In conclusione, gli organi regolatori e le società di consulenza, pur avendo ruoli e responsabilità distinti, lavorano in un ecosistema interconnesso. Le agenzie regolatorie stabiliscono le regole del gioco, mentre le società di consulenza aiutano le aziende a giocarlo al meglio. Questa collaborazione, anche se indiretta, è fondamentale per l’efficienza e la sostenibilità del sistema sanitario, garantendo che i pazienti abbiano accesso a terapie sicure ed efficaci nel minor tempo possibile, e al Sistema Sanitario di poter ottimizzare i processi e le economie sottostanti l’introduzione delle continue innovazioni tecnologiche. L’integrazione dei saperi e la possibilità di uno scenario multidisciplinare trova come luogo di possibile proprio la Direzione medica degli ospedali.

Bibliografia normativa e di riferimento
- Direttiva 2009/41/CE del Parlamento Europeo e del Consiglio del 6 maggio 2009, sull’impiego confinato di microrganismi geneticamente modificati.
- Decreto Legislativo 12 aprile 2001, n. 206 – Attuazione della direttiva 98/81/CE relativa all’impiego confinato di microrganismi geneticamente modificati.
- Linee guida del Ministero della Salute sull’impiego confinato di microrganismi geneticamente modificati e criteri di valutazione del rischio.
- Documenti e circolari dell’Istituto Superiore di Sanità (ISS) in materia di biosicurezza e biotecnologie.
- Comitato Nazionale per la Biosicurezza, le Biotecnologie e le Scienze della Vita (CNBBSV) – Relazioni e raccomandazioni.

Comments